


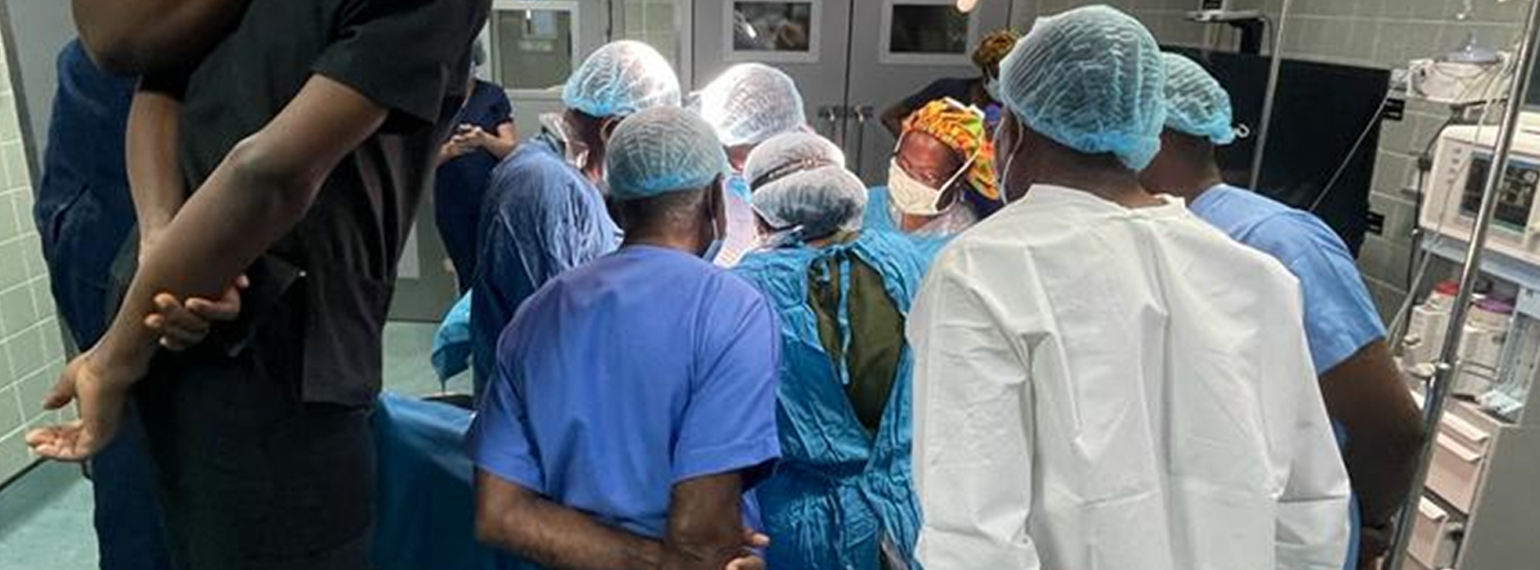

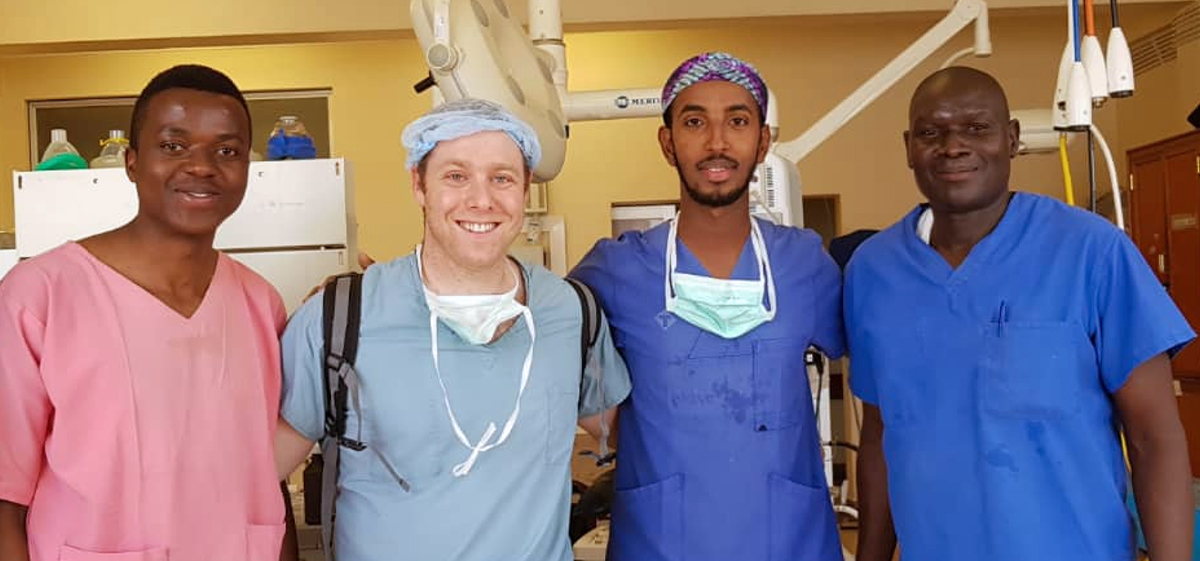
In all, 34 University of Michigan Medical School students embarked on educational experiences abroad in 2023, many with financial support from Global REACH. We support a variety of clinical and research-based programs for students seeking to engage in global health.
All medical students engaging in international travel for clinical electives and/or research field work must obtain authorization from Global REACH. Learn more.






